Brought to you by:
McGill Univ. Libraries and the generosity of Dr. Richard L. Tomlinson
McGill Univ. Libraries and the generosity of Dr. Richard L. Tomlinson
|
Brought to you by: McGill Univ. Libraries and the generosity of Dr. Richard L. Tomlinson |
||||
|
|
|
| Bioorganic
& Medicinal Chemistry Letters Volume 17, Issue 15, 1 August 2007, Pages 4248-4253 |
| ||||
|
|
|
| |
Optimization of novel combi-molecules: Identification of balanced and mixed bcr-abl/DNA targeting properties
Zakaria Rachidb,
†,
Athanasia Katsoulasb,
†,
Christopher Williamsa,
Anne-Laure Larroqueb,
James McNameeb
and Bertrand J. Jean-Claudeb,
![]() ,
, ![]()
Abstract
Steps toward the identification of combi-molecules with strong abl tyrosine
kinase (TK) inhibitory property and significant DNA damaging potential are
described. The optimized combi-molecule 13a was shown to induce
approximately twofold stronger abl TK inhibitory activity than Gleevec™ and high
levels of DNA damage in chronic myelogenous leukemic cells.
Graphical abstract
We designed and synthesized an optimized combi-molecule that induced
approximately twofold stronger abl TK inhibitory activity than Gleevec and high
levels of DNA damage in chronic myelogenous leukemic cells (CML).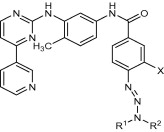
Keywords: bcr-abl; CML; Triazene; Inhibitors; Chronic
myelogenous leukemia; Pyridopyrimidine; Gleevec; Combi-molecule
The combi-targeting concept is a novel approach to drug design that seeks to synthesize compounds termed ‘combi-molecules’ capable of binding to kinases involved in cell signaling pathways while remaining DNA reactive.[1], [2], [3], [4], [5], [6], [7], [8], [9] and[10] This principle was developed on the premise that cancer cells are endowed with multiple mechanisms to overcome cytotoxic lesions. By inducing a tandem DNA damage and blockade of kinases that control signaling pathways related to the repair of the lesions in the cells, it was expected that the latter could be less prone to evade the death pathways. The chronic myelogenous leukemia (CML) is one of the diseases in which one such tyrosine kinase bcr-abl is known to be involved in two major cytoprotective events: (a) induction or activation of DNA repair proteins (e.g., Rad51),[11] and [12] (b) activation of the anti-apoptotic PI3K/AKT pathway.[13] and [14] Thus, we surmised that combi-molecules designed to block bcr-abl would on one hand alleviate anti-apoptotic signaling and on the other inflict cytotoxic DNA lesions: a strategy designed to induce enhanced cell-killing in CML cells.
Recently we reported on the synthesis of the first models to demonstrate the feasibility of the approach by designing two types of molecules: (1) those requiring hydrolytic cleavage to generate the DNA alkylating moiety termed ‘type I combi-molecules’,[8], [9] and [10] (2) those that can damage DNA without requirement for hydrolytic cleavage (type II). Unfortunately, the first probes synthesized to demonstrate the feasibility of the approach showed disproportionate bcr-abl-DNA targeting properties. The compound 2 (Scheme 1), a type II combi-molecule, while being a potent bcr-abl TK inhibitor (although less potent than Gleevec™) was a weak DNA damaging compound.5 The compound 13e (Scheme 2), a type I combi-molecule, while capable of inducing high levels of DNA damage was a moderate bcr-abl TK inhibitor.[4] and [7] Thus, to demonstrate the advantages of this novel approach, a structure optimized to show strong bcr-abl inhibitory potency and fierce DNA damaging potential was urgently needed. Here we report on the steps toward the identification of one such structure 13a, a triazene with balanced and enhanced bcr-abl-DNA targeting properties.
Display Full Size version of this image (40K) Scheme 1. Reagents and conditions: (i) THF/DIPEA (2 equiv)/0 °C; (ii) DMF/KI (1.1 equiv) at room temperature; (iii) POCl3/heat.
Display Full Size version of this image (32K) Scheme 2. Reagents and conditions: (i) HClconcd/heat; (ii) SOCl2, THF/heat; (iii) pyridine(2 equiv)/CH2Cl2/0 °C; (iv) Fe/ethanol/AcOH/heat; (v) CH3CN/NOBF4/−5 °C/ether then addition of alkylamine correspondent or 1:30 mixture of aqueous methylamine (40%) and aqueous formaldehyde (37%).
We first attempted to improve the DNA damaging potential of the type II combi-molecule 2 (because of its superior abl TK inhibitory activity) by inserting a CH2 group between its mustard moiety and the benzamide function (see 6, Scheme 1). This is inspired by the fact that the electron density at the nitrogen of 2 is depleted by the benzene ring, thereby reducing its alkylating power. Thus, we surmised that inserting a CH2 group in this part of the molecule would lead to a compound with not only a strong alkylating power, but also because its structure straddles that of Gleevec™, a strong bcr-abl inhibitory property. The synthesis of compounds 2, 5, and 6 proceeded according to Scheme 1. Briefly, amine 1, synthesized as previously described,15 was treated with p-{N,N-bis(2-chloroethyl)amino}benzoyl chloride16 or p-{N,N-bis[(2-chloroethyl)aminomethyl]}benzoyl chloride17 to give compounds 2 or 6. Treatment of 1 with 4-chloromethylbenzoyl chloride gave 3 which was stirred in DMF with N-methylethanolamine to provide 4. Chlorination of amino-alcohol 4 with POCl3 gave the hemi-mustard 5 in good yield. Compound 5 was a poor bcr-abl TK inhibitor and inactive against K562 cell. Likewise, compound 6 was more than 50-fold less potent than Gleevec™ and virtually inactive against the CML cell line K562 (Table 1).
Cytotoxicity and Abl tyrosine kinase inhibitory activities by different compounds
a IC50 values were determined by using the c-abl tyrosine kinase protein. Values are means of two separate experiments.
Compound Inhibition of abl kinase IC50,a μM IC50,b μM (K562) Gleevec™ 0.04 0.18 25 0.22 2.46 5 3.11 79.72 6 >10 >100 11b 0.156 0.316 11a >10 68 13a 0.028 0.113 13b 0.018 0.052 13c 0.033 0.187 13d 0.180 9.98 13e7 1.057 3.15
b IC50 values were obtained from the MTT assay.
Molecular modeling was used to account for the failure of compound 6, the design of which was simply based on structural overlap with Gleevec™. As depicted in Figure 1, the phenyl ring of compound 2 (Scheme 1) is anchored into a hydrophobic pocket of bcr-abl, allowing its mustard moiety to occupy a region in the receptor where minimum steric clash can occur. In contrast, the CH2 group of 6 protrudes the mustard moiety into an orientation where its steric bulk can induce clash with the pocket. Reducing the size of the mustard by creating a hemi-mustard 5 (Scheme 1) resulted in a compound with greater affinity for bcr-abl but with a potency approximately 75-fold less than that of Gleevec™. Thus the type II strategy involving the bulky mustard group was abandoned. However, data acquired from this work allowed us to better understand the mode of binding of the combi-molecules, most particularly the anchorage of the benzamide ring into a narrow hydrophobic pocket and the requirement for a non-bulky DNA damaging tail. For the latter property, the triazene tail already demonstrated to induce high levels of DNA damage in the pyrido-pyrimidine system could now be selected as the group of choice.7 However, for enhancing the interaction of the abl recognition moiety, the challenge remained.
Display Full Size version of this image (25K) Figure 1. Compounds 2 and 6 in 1IEP (X-ray structure of Gleevec™ shown in yellow).
Based upon results obtained for 2, we thought it of interest to enhance hydrophobic interactions of the benzamide moiety with the hydrophobic pocket. Interestingly, during the course of this development, Asaki et al.,18coincidentally addressing the same issue, demonstrated that compounds with small hydrophobic substituents, for example, Cl and CF3 on the benzamide moiety could show enhanced binding affinity when compared with Gleevec™. Thus inspired by our results on 2 and the latter development, we designed combi-molecules containing: (a) a Cl and CF3 group on the phenyl ring of the benzamide moiety, and (b) a small 1,2,3-triazene tail carrying one or two methyl groups at the 3-position.
The synthesis of compounds 13a–d proceeded according to an adaptation of a strategy developed for the synthesis of 13e.7 Briefly, commercially available compound 7 was hydrolyzed under acidic condition to give the acid 8 which was chlorinated with SOCl2 to give the benzoyl chloride 9. Treatment of 1 with the benzoyl chloride 9 in methylene chloride/pyridine (2 equiv) at 0 °C gave the nitro compounds 10, which were reduced with Fe in ethanol to provide the amines 11a–b, and 12. Diazotization of these amines with nitrosonium tetrafluoroborate in acetonitrile followed by the addition of the desired amine and neutralization with triethylamine gave triazene conjugates 13a–d.19
Interestingly, abl TK enzyme assays showed that compound 13d containing a 3-Cl group showed potency similar to that of 2 but 10-fold better than 13e. The combi-molecules with the more hydrophobic CF3 group, 13a–c, showed abl TK inhibitory potency not only superior to that of 13e but also to that of Gleevec™. Compound 13b, the dimethyl analog of 13a, was the most potent of the series, which we believe is due to its more hydrophobic dimethyl function. It should be noted that the latter compound could not be considered to be a combi-molecule in vitro since like all dimethyl-triazenes,[20] and [21] it cannot be converted to 13a without metabolic activation.
One of the special features of type I combi-molecules is their ability to not only block their target on their own but also to further degrade into another metabolite with bcr-abl TK inhibitory activity that sustains inhibition of bcr-abl TK. HPLC analysis showed that more than 70% of 13a was degraded to 11b as early as 3 h after incubation in cell culture medium at 37 °C. It should be remembered here that the previous non-optimized combi-molecule 13e degraded to generate the amine 11a which was found to be virtually inactive against bcr-abl TK-expressing cells.[4] and [7] In contrast, compound 11b is 400-fold more potent than the latter amine and only threefold less potent than Gleevec™. Molecular modeling study suggests that the superior potency of 11b may be mediated as for the combi-molecule by the ability of the CF3 group to occupy a hydrophobic subpocket in the ATP binding site of the receptor (Fig. 2). The 1IEP crystal structure of Abl kinase bound to Gleevec22 was used as a starting point to construct compounds 13 analogs in the Abl pocket. The 1IEP PDB file was loaded into the MOE software package23; all crystallographic water molecules were deleted and hydogen atoms were added. The ligand coordinates of Gleevec™ from the 1IEP X-ray structure were used as a template to manually construct starting structures for the compounds 13 compounds in the pocket of the 1IEP crystal structure. MMFF94x[23] and [24] partial charges were assigned to all the atoms in the system, and each ligand structure was minimized to a gradient of 0.01 kcal/Å mol in the presence of the receptor using the MMFF94x forcefield in MOE. The receptor atoms were held fixed throughout all the minimizations. In all cases, the structures minimized to coordinates very similar to the Gleevec™ X-ray coordinates. Furthermore, the minimizations show that there is sufficient room for the CF3 substituent in the hydrophobic subpocket. Experimental evidence for occupation of this pocket is provided by the 2HIW PDB crystal structure of complexed abl kinase, which shows a CF3 substituent on the inhibitor occupying this subpocket.25 For the optimized combi-molecule 13a, Poisson–Boltzmann electrostatic maps of the entire abl pocket26 (Fig. 3) show a 3 kcal/mol hydrophobic isocontour in the subpocket, suggesting that hydrophobic contact with this region will yield an approximate 3 kcal/mol gain in ligand binding energy. Indeed, this predicted 3 kcal/mol binding energy gain correlates well (Table 2) with the increase in affinity observed between pairs of compounds that differ only by CF3 substituent [11a/11b and 13e/13c].
Display Full Size version of this image (35K) Figure 2. Occupation of 1IEP subpocket by ortho CF3 group of 11b.
Display Full Size version of this image (33K) Figure 3. Compound 13a docked in 1IEP abl pocket. Green surfaces reflect the 3 kcal/mol electrostatic energy isocontour for hydrophobic contacts within the pocket.
Table 2.Binding energy gains from –CF3 substitution on benzamide moiety in Gleevec™ analogs
a Binding energy gains estimated from experimental IC50 values using ΔΔG = −rt ln (IC50CF3/IC50H)27.
Compound pair Inhibition of abl kinase IC50, μM ΔΔGa (kcal/mol) X = –H X = CF3 11a/11b 67 0.156 3.6 13e/13c 1.1 0.033 2.1
In order to determine the translation of the now proportionate binary targeting property of the combi-molecules, we studied their potency against the bcr-abl expressing CML cells K562. The results showed that 13a is twofold more potent than Gleevec™ and fivefold more potent than 11b. Furthermore, its DNA damaging potential was demonstrated by the comet assay. Interestingly, compound 13a induced significant levels of DNA damage in the Mo7p210 transfected cells (Fig. 4).
Display Full Size version of this image (5K) Figure 4. Quantitation of DNA damage using the alkaline comet assay. DNA damage induced by 13a, 11b and Temozolomide (TEM) in the M07/p210 cell line. Tail moment was used as a parameter for the detection of DNA damage in M07/p210 cells exposed to the drugs 30 min.
This study conclusively demonstrated that the potency of the combi-molecules
could be refined by enhancing the hydrophobicity of the benzamide triazene
carrier with a binding mode similar to that of Gleevec™. The now augmented abl
TK inhibitory potency (activity superior to Gleevec™) has conferred to these
Type I combi-molecules of the triazene class, strong and balanced bcr-abl/DNA
targeting properties. To our knowledge, this is the first report on such a
combi-molecule with balanced bcr-abl/DNA targeting properties. A complete study
on its biological mechanism of action (blockade of abl phosphorylation in whole
cells, analysis of the DNA damage response pathways p53, p21, induction of
apoptosis (Bax), and comparative analysis with individual combinations of
bcr-abl + DNA damaging agents will be reported elsewhere.
Acknowledgments
We thank the Leukemia and Lymphoma society for financial support. A. Katsoulas is grateful to the Fonds de la Recherche en Santé du Québec (FRSQ) for a doctoral studentship.
References and notes
1 Q. Qiu, J. Domarkas, R. Banerjee, N. Merayo, F. Brahimi, J.P. McNamee, B.F. Gibbs and B.J. Jean-Claude, Clin. Cancer Res. 13 (2007), p. 331. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (2)
2 Q. Qiu, J. Domarkas, R. Banerjee, A. Katsoulas, J.P. McNamee and B.J. Jean-Claude, Anti-Cancer Drugs 18 (2007), p. 171. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (1)
3 J. Domarkas, F. Dudouit, C. Williams, Q. Qiyu, R. Banerjee, F. Brahimi and B.J. Jean-Claude, J. Med. Chem. 49 (2006), p. 3544. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (4)
4 A. Katsoulas, Z. Rachid, F. Brahimi, J. McNamee and B.J. Jean-Claude, Leuk. Res. 29 (2005), p. 693. SummaryPlus | Full Text + Links | PDF (412 K) | View Record in Scopus | Cited By in Scopus (2)
5 A. Katsoulas, Z. Rachid, F. Brahimi, J. McNamee and B.J. Jean-Claude, Leuk. Res. 29 (2005), p. 565. SummaryPlus | Full Text + Links | PDF (227 K) | View Record in Scopus | Cited By in Scopus (1)
6 R. Banerjee, Z. Rachid, J. McNamee and B.J. Jean-Claude, J. Med. Chem. 46 (2003), p. 5546. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (16)
7 Z. Rachid, A. Katsoulas, F. Brahimi and B.J. Jean-Claude, Bioorg. Med. Chem. Lett. 13 (2003), p. 3297. SummaryPlus | Full Text + Links | PDF (168 K) | View Record in Scopus | Cited By in Scopus (2)
8 S.L. Matheson, J.P. McNamee and B.J. Jean-Claude, Cancer Chemother. Pharmacol. 51 (2003), p. 11. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (19)
9 Q. Qiu, F. Dudouit, S.L. Matheson, F. Brahimi, R. Banerjee, J.P. McNamee and B.J. Jean-Claude, Cancer Chemother. Pharmacol. 51 (2003), p. 1. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (19)
10 F. Brahimi, S.L. Matheson, F. Dudouit, J.P. McNamee, A.M. Tari and B.J. Jean-Claude, J. Pharmacol. Exp. Ther. 303 (2002), p. 238. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (22)
11 A. Slupianek, C. Schmutte, G. Tombline, M. Nieborowska-Skorska, G. Hoser, M.O. Nowicki, A.J. Pierce, R. Fishel and T. Skorski, Mol. Cell 8 (2001), p. 795. SummaryPlus | Full Text + Links | PDF (568 K) | View Record in Scopus | Cited By in Scopus (104)
12 N. Takeda, M. Shibuya and Y. Maru, Proc. Natl. Acad. Sci. U.S.A. 96 (1999), p. 203. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (38)
13 K. Kawauchi, T. Ogasawara, M. Yasuyama and S.-i. Ohkawa, Blood Cells Mol. Dis. 31 (2003), p. 11. SummaryPlus | Full Text + Links | PDF (330 K) | View Record in Scopus | Cited By in Scopus (17)
14 T. Skorski, A. Bellacosa, M. Nieborowska-Skorska, M. Majewski, R. Martinez, J.K. Choi, R. Trotta, P. Wlodarski, D. Perrotti, T.O. Chan, M.A. Wasik, P.N. Tsichlis and B. Calabretta, EMBO J. 16 (1997), p. 6151. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (347)
15 J. Zimmermann, E. Buchdunger, H. Mett, T. Meyer and N.B. Lydon, Bioorg. Med. Chem. Lett. 7 (1997), p. 187. Abstract | Abstract + References | PDF (279 K) | View Record in Scopus | Cited By in Scopus (87)
16 R.C. Elderfield and T.-K. Liao, J. Org. Chem. 26 (1961), p. 4996. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (11)
17 Hatono, S.; Yazaki, A.; Yokomoto, M.; Hirao, Y. 87-307846 259185, 1988, 26 pp.
18 T. Asaki, Y. Sugiyama, T. Hamamoto, M. Higashioka, M. Umehara, H. Naito and T. Niwa, Bioorg. Med. Chem. Lett. 16 (2006), p. 1421. SummaryPlus | Full Text + Links | PDF (201 K) | View Record in Scopus | Cited By in Scopus (6)
Experimental data for 13a.
1-{N-[4-Methyl-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-phenyl]-3-tri
fluoromethyl-benzamide}-methyl triazene. The amine 11b
(100 mg, 0.22 mmol) was dissolved in dry acetonitrile (10 mL) and
after cooling to −5 °C, a solution of nitrosonium tetrafluoroborate
(50 mg, 0.5 mmol) suspended in acetonitrile was added. The clear
solution was stirred for 1 h at −5 °C and a mixture of ether
(10 mL), water (2 mL), triethylamine (0.43 mL) and aqueous
methylamine (40%) (0.2 mL) was added dropwise. The mixture was subsequently
stirred at 0 °C for 15 min, the reaction was extracted with ethyl
acetate dried then evaporated and the precipitate that formed triturated and
washed with ether to give 13a (50 mg, 42%) as a pure
solid. ESI m/z 529.0 (MNa)+; 1H NMR (400 MHz,
DMSO-d6) δ 11.30 (br s, 1H, NN–NMeH), 10.33 (s, 1H, NH), 9.27 (s, 1H,
ArH), 8.99 (s, 1H, ArH), 8.67 (d, 1H, J = 4.8 Hz, ArH),
8.51–8.46 (m, 2H, ArH), 8.26 (s, 1H, ArH), 8.18 (d, 1H,
J = 12.4 Hz, ArH), 8.06 (s, 1H, ArH), 7.64 (d, 1H,
J = 11.2, J = 11.2 Hz, ArH), 7.53–7.42
(m, 3H, ArH), 7.21 (d, 1H, J = 11.2, ArH), 3.07 (s, 3H,
NHCH3), 2.22 (s, 3H, CH3); 13C NMR
(75 MHz, DMSO-d6) 168.7, 164.5, 162.3, 161.9, 160.2,
152.1, 151.8, 148.9, 138.5, 138.4, 137.7, 135.1, 133.2, 132.9, 131.4, 130.8,
128.5, 126.6, 124.5, 118.1, 118.0, 117.6, 108.2, 31.4, 18.3.
20 H.W. Manning, L.M. Cameron, R.J. LaFrance, K. Vaughan and R. Rajaraman, Anti-Cancer Drug Des. 1 (1985), p. 37. View Record in Scopus | Cited By in Scopus (11)
21 L.M. Cameron, R.J. LaFrance, C.M. Hemens, K. Vaughan, R. Rajaraman, D.C. Chubb and P.M. Goddard, Anti-Cancer Drug Des. 1 (1985), p. 27. View Record in Scopus | Cited By in Scopus (14)
22 B. Nagar, W.G. Bornmann, P. Pellicena, T. Schindler, D.R. Veach, W.T. Miller, B. Clarkson and J. Kuriyan, Cancer Res. 62 (2002), p. 4236. View Record in Scopus | Cited By in Scopus (272)
23 MOE software (Version 2006.07) available from Chemical Computing Group Inc., S.S.W., Montreal, Quebec, Canada. http://www.sciencedirect.com/science?_ob=RedirectURL&_method=externObjLink&_locator=url&_plusSign=%2B&_targetURL=http%253A%252F%252Fwww.chemcomp.com.
24 T.A. Halgren, J. Comput. Chem. 20 (1999), p. 720. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (188)
25 B. Okram, A. Nagle, F.J. Adrian, C. Lee, P. Ren, X. Wang, T. Sim, Y. Xie, X. Wang, G. Xia, G. Spraggon, M. Warmuth, Y. Liu and N.S. Gray, Chem. Biol. 13 (2006), p. 779. SummaryPlus | Full Text + Links | PDF (439 K) | View Record in Scopus | Cited By in Scopus (9)
Non-linear Poisson–Boltzmann calculation as implemented in the MOE software package (Version 2006.08) available from Chemical Computing Group Inc.; 1010 Sherbrooke St. West, M., Quebec, Canada http://www.sciencedirect.com/science?_ob=RedirectURL&_method=externObjLink&_locator=url&_plusSign=%2B&_targetURL=http%253A%252F%252Fwww.chemcomp.com.
27
The Practice of Medicinal Chemistry, 2nd ed.; Wermuth, C. G., Ed.; 2003.
| Bioorganic
& Medicinal Chemistry Letters Volume 17, Issue 15, 1 August 2007, Pages 4248-4253 |
|
|
 |
 - Opens new window")
.gif "Display Full Size version of this image (32K) - Opens new window")
.gif "Display Full Size version of this image (25K) - Opens new window")
.gif "Display Full Size version of this image (35K) - Opens new window")
.gif "Display Full Size version of this image (33K) - Opens new window")
.gif "Display Full Size version of this image (5K) - Opens new window")